





 3597
3597(Suite de la page Cancer - sources)
Avertissement :
Je n’ai ni les compétences ni le temps d’étudier les nombreuses annonces (ou promesses) de nouveaux traitements du cancer relayées par les médias avec un enthousiasme compréhensible. Ce qui suit n’est donc que le suivi de dossiers anciens sur lesquels ont été publiés des commentaires ou des critiques accessibles au lecteur non-spécialiste…
Une autre raison de cette réserve est que la majorité des articles qui ont fait l’objet de falsifications — manipulations d’images détectées par des outils récents — relèvent des domaines de la cancérologie ou de disciplines connexes. J’ai mentionné ce fait sur la page Ma démarche. La recherche sur le cancer drainant des ressources financières considérables, il est tentant pour un chercheur ou une équipe d’en faire apparaître les résultats sous un jour « positif », en contradiction avec les « données brutes » produites par leurs travaux.
Sommaire
⇪ Un recadrage des priorités
James Dewey WatsonN1, lauréat du Prix Nobel en 1962 et codécouvreur avec Francis Crick de la structure de l’ADN (sur la base du travail déterminant mais scandaleusement passé sous silence de Rosalind FranklinN2) a défrayé la chronique en publiant dans The Lancet (février 2014N3) une hypothèse selon laquelle le diabète de type 2, les formes de démence, les maladies cardiovasculaires et certains cancers seraient causés par l’incapacité de produire suffisamment des dérivés réactifs de l’oxygène (ROSN4) — aussi appelés « radicaux libres ».
Il ajoutait qu’une meilleure compréhension des effets bénéfiques de l'exercice physique de haute intensité (HIITN5) pourrait contribuer à soigner cette incapacité. Peter Tarr commente (2014N6) :
Il ne remet pas en question le fait que le tissu pancréatiqueN7 est enflammé chez les patients du diabète de type 2N8. Mais il propose une théorie explicative nouvelle : « Je suggère que la cause principale est une déficience d’agents oxydantsN4 et non un excès. »
Watson rappelle que les cellules du corps ne peuvent pas survivre sans fabriquer à la fois des agents oxydants et antioxydants. Il existe un équilibre subtil entre les deux. L’exercice physique (de haute intensité) oblige le corps à fabriquer un grand nombre d’agents oxydants : ROSN4 ou « radicaux libres ». Dans le réticulum endoplasmiqueN10, une des espèces de ROS, le peroxyde d’oxygène H2O2 sert à créer des liens chimiques (ponts disulfureN11) stabilisant les protéines lorsqu’elles se replientN12. Si l’oxydation est insuffisante, les protéines ne se replient pas et deviennent inactives. C’est, selon Watson, ce qui cause l’inflammation du pancréas conduisant au diabète de type 2N8. Il note que les athlètes qui absorbent une grande quantité d’antioxydants avant l’effort auraient tendance à diminuer son effet bénéfique. Cette proposition avait été vérifiée par Michael Ristow et collègues (2009N13) — voir aussi la mito-hormèseN14 selon Michael Ristow & Kathrin Schmeisser (2014N15).
Dans son article Oxidants, antioxidants and the current incurability of metastatic cancers (2013N16), James Watson développait le même type d’argument pour ce qui concerne le cancer.
Nous devrions nous concentrer beaucoup, beaucoup plus, sur l’éventail très large de vulnérabilités métaboliques et oxydatives qui apparaissent en conséquence des capacités incontrôlables de croissance et de prolifération des cellules cancéreuses. Lorsque les cancers humains dérivent vers des états glycolytiques plus agressifs, leur stress métabolique en augmentation constante les rend particulièrement vulnérables à une baisse soudaine de leurs fournitures vitales d’énergie sous la forme d’ATPN17.
Les cellules tumorales d’origine épithéliale (carcinomesN18) peuvent subir une transition épithélio-mésenchymateuse (TEMN19) qui les rend invasives et constitue une des premières étapes vers la formation de métastasesN20. L’auteur explique que la TEM préserve l’agencement initial des bases de l’ADN tout en modifiant la manière dont elles sont transcrites en ARN. La régulation de cette transcription influence la réponse des cellules cancéreuses à leur environnement. Elle est du même ordre que celle qui permet la transition des œufs fertilisés vers les cellules différenciées (poumon, rein etc.) dans les organismes adultes.
Selon Watson, il serait préférable de chercher en priorité des médicaments susceptibles de bloquer la prolifération des cellules cancéreuses, plutôt que leur croissance, car ces derniers ont un effet indésirable sur les cellules saines. Plusieurs pistes de recherche sont suggérées — que je ne sais pas analyser… — notamment pour désinhiber le mécanisme d’apoptose (N21 mort cellulaire programmée) assurant l’élimination des cellules endommagées. La production d’agents oxydants (ROSN4) est un des activateurs de cette apoptose, comme l’a prouvé le succès du médicament mitochrondrial elesclomol (de Synta Pharmaceuticals), mais cette production est aussi, nous l’avons vu, un des effets bénéfiques de l’exercice de haute intensité (N5). Selon Watson (2013N16),
Quand les molécules de ROS ainsi formées sont détruites par l’administration simultanée d’une molécule anti-oxydante N‑acétylcystéineN22, la destruction ciblée des cellules cancéreuses s’interrompt. Le fait que l’elesclomol ne peut pas déclencher l’apoptose des cellules non-cancéreuses est probablement dû au plus bas niveau de ROSN4 intrinsèquement généré par la machinerie mitochondriale normale de transport des électrons.
Il semblerait pour lui que l’efficacité de certains traitements de chimiothérapie, aussi bien que celle de rayonnements ionisants, tienne à leur faculté de produire des radicaux libres (ROSN4) utilisés pour l’apoptoseN21 des cellules endommagées. Ce qui pourrait expliquer que certains cancers devenus résistants à la chimiothérapie résistent aussi à la radiothérapie. Les cellules cancéreuses contrôlées principalement par les oncogènesN23 RAS et Myc, qui apparaissent notamment en phase terminale, sont parmi les plus difficiles à éliminer. L’auteur suggère que cela pourrait être dû à leur production importante d’anti-oxydants destructeurs de radicaux libres (ROS), notamment le glutathionN24, les superoxyde dismutasesN25, la catalaseN26 et la thiorédoxyneN27, production contrôlée par le facteur de transcription Nrf2N28 ; ce facteur est à son tour contrôlé par les oncogènes RAS, RAF et Myc qui favorisent la croissance et la division des cellules.
Watson rappelle enfin que de nombreuses études sur la supplémentation en anti-oxydants, principalement le bétacarotène, les vitamines A, C, E et le sélénium, n’ont montré aucune efficacité dans la prévention du cancer gastro-intestinal ni dans la diminution de la mortalité. « Au contraire, il semble qu’ils réduisent légèrement l’espérance de vie de ceux qui les consomment ».
Pour ce qui concerne l’exercice de haute intensité (N5), on entend parfois dire qu’en favorisant la production d’hormone de croissance il entraînerait la croissance des cellules cancéreuses. En réalité, c’est le facteur de croissance ressemblant à l’insuline (IGF‑1N29) qui en est responsable. L’hormone HGH produit de l’IGF‑1 (par l’intermédiaire du foie), mais elle produit aussi des récepteurs de l’IGF‑1 qui contrecarrent son effet sur les cellules cancéreuses. Le rapport entre HGH et IGF‑1 est décrit dans l’article Regulation of muscle mass by growth hormone and IGF‑1 (Velloso CP, 2008N30).
Watson (2013N16) décrivait ce qu’il percevait comme une impasse dans la poursuite de la recherche sur le cancer :
Les thérapies génétiques individuelles du cancer dont on fait grand cas en ce moment pourraient s’avérer beaucoup moins intéressantes pour la médecine à venir que ne le laissent penser les articles de presse aujourd’hui. Si l’on attribuait plus de fonds de recherche gouvernementaux sur le cancer au développement de médicaments nouveaux anti-métastasiques par les institutions académiques de haut niveau bien choisies, les fonds du National Cancer Institute (NCI) seraient mieux utilisés que les larges sommes dépensées à essayer des médicaments qui ont peu de chance d’apporter du nouveau. Le plus grand obstacle aujourd’hui à une véritable guerre contre le cancer peut être attribué au caractère intrinsèquement conservateur des établissements de recherche sur le cancer. Ils sont encore trop étroitement attachés à aller de l’avant avec des cocktails de médicaments ciblant les molécules (comme HER2, RAS, RAF, MEK, ERK, PI3K, AKT et mTOR) qui facilitent la croissance des voies de transduction du signal, au lieu de cibler les molécules Myc qui gouvernent spécifiquement le cycle cellulaire.
[…]
Au sommet du grand budget de la science au National Cancer Institute, on aperçoit encore le projet The Cancer Genome Atlas (TCGA), qui par nature ne découvre que les déclencheurs de cancer au détriment des vulnérabilités (partenaires synthétiquement mortels). Bien qu’au début j’aie soutenu TCGA dans sa recherche d’aides financières importantes, je ne le fais plus à présent. Les 100 millions de dollars injectés chaque année ont peu de chance de produire les médicaments vraiment de pointe dont nous avons tant besoin.
Dans le même sens, Andrew Porterfield déplorait que la « guerre contre le cancer » déclarée en 1971 par le Président Richard Nixon soit actuellement focalisée sur la recherche génétique (2015N31) :
Un problème pourrait être le fait que les cancers à tumeurs qui apparaissent en premier dans le corps, identifiables par leur emplacement, que ce soit le foie, le poumon, le cerveau ou le colon, ne sont pas les causes principales de décès par cancer. La plupart des gens meurent à cause des cellules cancéreuses qui se détachent des tumeurs primaires et s’installent dans d’autres parties du corps. Ce mécanisme de métastaseN20 est responsable de 90% des décès par cancer. Or seulement 5% des fonds de recherche européens sur le cancer, et 2% aux USA, sont consacrés à la recherche sur les métastases.
James Watson concluait son article (2013N16) par l’espoir que les grandes firmes pharmaceutiques entreprendraient une étude concertée, au moins sur les principaux cancers (sein, colon et poumon), en exploitant la technique d’interférence par ARN (RNAiN32). Cette technique suscitait de grands espoirs (voir Kaelin WG, 2012N33) et sa mise en œuvre nécessitait un investissement de moins d’un milliard de dollars. Un projet ciblant la totalité du génome a été initié par Pfizer en collaboration avec le Cold Spring Harbor LaboratoryN34.
La proposition de Watson en 2009 (To Fight Cancer, Know the EnemyN35) — changer son fusil d’épaule dans la recherche de traitements du cancer, par un retour sur scène de la théorie métabolique — pourrait avoir été inspirée par Lewis Cantley, fondateur de la start-up AgiosN36 focalisée sur le métabolisme du cancer. En effet, selon Christofferson (2014N37, p. 119–120), James Watson aurait transmis à Cantley le dossier qui lui avait été confié par Young Ko, à titre confidentiel, sur l’expérimentation du traitement par 3BP (voir ci-dessous). Watson déclare :
L’idée que les cellules cancéreuses puissent partager un ensemble commun de molécules qu’on ne trouve pas dans la plupart des autres cellules de notre corps a été proposée en premier par le grand biochimiste allemand Otto Warburg. En 1924, il a observé que toutes les cellules cancéreuses, indépendamment du fait qu’elles croissent en la présence ou en l’absence d’oxygène, produisent de grandes quantités d’acide lactique. Mais c’est seulement il y a un an que le sens de la découverte de Warburg a été révélé : le métabolisme des cellules cancéreuses, et bien sûr de toutes les cellules qui prolifèrent, est largement dirigé vers la synthèse des blocs de construction cellulaires à partir des produits de dégradation du glucose.
James Watson (2009N35)
En sous-entendant que l’hypothèse de WarburgN38 aurait été réhabilitée « il y a un an », autrement dit par Cantley et son équipe, Watson faisait l’impasse sur plus de trente années de travaux effectués par Peter Pederson dans la continuation de ceux de Warburg — mais il semble être coutumier du procédé vu le sort qu’il a réservé à Rosalind FranklinN39 !
⇪ Warburg, le retour
En 2000, Douglas Hanahan et Robert Weinberg ont publié dans Cell un article majeur — le plus cité de cette revue — qui fait le point sur six propriétés caractéristiques des cancers (The Hallmarks of CancerN40) : les cellules cancéreuses stimulent leur propre croissance, elles sont insensibles aux signaux inhibiteurs de la croissance, elles peuvent éviter la mort cellulaire programmée (apoptoseN21), elles ont la capacité de se reproduire indéfiniment, elles induisent la capacité à faire croître de nouveaux vaisseaux sanguins qui permettent la croissance des tumeurs (angiogenèseN41) et elles se répandent sur des sites distants (métastasesN20).

Source : N42
En mars 2009, Peter Pedersen a été invité à intervenir dans un séminaire au NIH (voir vidéoN43) au cours duquel il a parlé de la découverte du 3BP (voir ci-dessous), mais aussi évoqué l’hypothèse de Hanahan et Weinberg en signalant (10 minutes après le début de l’exposé) qu’ils avaient omis d’inclure l’effet WarburgN44 dans leur liste. Suite à cette intervention, Hanahan et Weinberg ont publié en 2011 un deuxième article, Hallmarks of cancer : the next generationN45, identifiant deux capacités distinctives émergentes : la dérégulation du métabolisme énergétique cellulaire et la capacité d’éviter une destruction par le système immunitaire (voir WikipediaN46).
Weinberg reconnaissait maintenant l’effet WarburgN44 mais sans l’associer à un dysfonctionnement des mitochondriesN47. Il le considérait comme provenant du noyau : une reprogrammation du métabolisme conduite par les oncogènes.

Source : N48
Thomas SeyfriedN49 professeur de biologie au Boston College (Université de l’Illinois), cite l’expérience de McKinnel RG et al. (1969N50) qui consistait à transplanter le noyau d’une cellule cancéreuse d’une grenouille en remplacement de celui d’une cellule saine de têtard : malgré l’altération génétique de son noyau, la nouvelle cellule n’a pas donné lieu à une prolifération cancéreuse (Seyfried T, 2015aN51 et 2015bN52).
Cette expérience met en défaut la théorie génétique du cancer (SMT) car elle montre que des mutations significatives pourraient avoir lieu dans le cytoplasme (i.e. les mitochondriesN47) plutôt que dans l’ADN du noyau cellulaire. Ces résultats ont montré que des noyaux provenant de cellules tumorales pouvaient diriger un développement normal et n’ont pas induit une croissance cellulaire dérégulée, le phénotype de signature de la tumorigenèse (Seyfried T, 2015aN51).
Seyfried a revisité la théorie d’Otto Warburg selon laquelle tout cancer serait en premier lieu l’effet d’un dérèglement de métabolisme cellulaire, en l’actualisant à partir des données de la biomédecine et de la compréhension récente du fonctionnement des mitochondries. Il cite des cas de cancer du cerveau comme preuves du bien-fondé de solutions métaboliques au traitement de la maladie ; il expose des similarités avec d’autres types de cancer, notamment du sein et du côlon, liées à l’identité de leurs mutations cellulaires. Toutefois, son expérimentation se limite au cas de tumeurs cérébrales sur des modèles animaux, ce qui limite fortement le niveau de preuve pour les autres cancers et l’applicabilité aux humains des traitements.
Michael O’Neill (2013bN53) commente :
Une des parties les plus magnifiquement écrites et convaincantes de l’hypothèse exhaustive de Seyfried est l’idée que la métastaseN20 est un processus trop complexe pour être pris en compte par des mutations génétiques aléatoires. L’idée que de nombreux types de cellules cancéreuses recueilleraient en quelque sorte les mutations génétiques correctes pour leur permettre d’entrer et de sortir des tissus, échapper à la détection par le système immunitaire, et se propager dans tout le corps semble ridicule. Dès les débuts de Cancer as a Metabolic Disease, Seyfried commence à remettre en question cela et montrer comment le processus de métastase implique des capacités déjà présentes dans certains macrophagesN54 et leucocytesN55. […] C’est un thème récurrent dans le livre, et à certains endroits il remarque même que « Aichel [1911*] a suggéré il y a près d’un siècle que la progression de la tumeur impliquait une fusion entre des leucocytes et des cellules somatiquesN56 ». Et c’est cette théorie de la fusion, et non des mutations génétiques, que Seyfried préconise comme source de la capacité d’une cellule cancéreuse à métastaser.
(*) Aichel O. (1911). About cell fusion with qualitatively abnormal chromosome distribution as cause for tumor formation. In : Roux W, editor. Vorträge und Aufsätze über Entvickelungsmechanik Der Organismen. Leipzig, Germany : Wilhelm Engelmann, 1911 : 92–111. (En allemand)
Cette hypothèse est confirmée par Rossitza Lazova et al. (2013N57) qui ont observé un mécanisme de fusion dans les métastases d’un cancer du cerveau consécutif à une greffe de moëlle épinière. John M Pawelek, un des co-auteurs de la publication, commente (Dodson H, 2013N58) :
Nos résultats constituent la première preuve pour les [cancers] humains d’une théorie proposée en 1911 par un pathologiste allemand [O Aichel], que la métastaseN20 se produit quand un leucocyteN55 et une cellule cancéreuse fusionnent en formant un hybride génétique. Cela pourrait ouvrir la voie à de nouvelles cibles thérapeutiques, mais beaucoup de travail reste à faire pour déterminer la façon dont la fusion se produit, la fréquence de ces hybrides dans les cancers humains, et le rôle potentiel des hybrides dans les métastases.
Pawelek écrit dans un autre article consacré à une expérimentation animale (2014N59):
Le modèle est simple : cellule blanche du sang + cellule cancéreuse non-métastasique = cellule cancéreuse métastasique. Mais il fournit une explication profonde et unificatrice de la métastaseN20.
Travis Christofferson (2014N37 p. xviii-seq) explique à sa manière le retour à une théorie métabolique du cancer :

Quand je me suis penché sur les données issues de TCGA, ce que j’ai découvert était étonnant. Rien ne faisait sens. Avant le projet, les chercheurs croyaient fermement que les données de séquençage révèleraient une séquence ordonnée de peut-être trois à huit gènes, qui, après avoir muté, se manifesteraient dans un type particulier de cancer — une signature comparable à une empreinte digitale — et qu’ils pourraient travailler à partir de cette signature pour développer des traitements. Mais ce que les données de séquençage ont révélé n’avait rien d’ordonné. Elles explosaient en une collection presque aléatoire de mutations — dont pas une seule, ni une configuration appropriée, était absolument responsable du déclenchement de la maladie.
[…]
Pourquoi les médicaments ciblés qu’on avait promis ne se sont-ils pas matérialisés ? Pour commencer, TCGA n’a pas réussi à identifier les mutations qui sans équivoque causaient un type donné de cancer. Par conséquent, les chercheurs n’ont pas pu trouver la ou les cibles correctes. Deuxièmement, une autre découverte a été faite grâce à TCGA, de celles qui projettent un nuage sombre sur tout espoir de percée significative dans l’avenir. D’un point de vue génétique, la conception du médicament est un jeu difficile et brutal de « attrape-moi si tu peux ». Les cibles mutationnelles n’étaient pas seulement très différentes d’une personne à une autre, elles variaient aussi spectaculairement d’une cellule à l’autre à l’intérieur de la même tumeur, confrontant les pharmacologues à une tâche de difficulté insurmontable.
[…]
Au lieu de cibler des mutations qui peuvent varier d’une seconde à l’autre, la théorie métabolique a remis les chercheurs sur le siège du conducteur. […] Bien que méconnues et dépréciées, les thérapies issues de la logique que le cancer provient d’un métabolisme endommagé ont donné des résultats remarquables. Les thérapies métaboliques découlent d’un simple agencement logique. Toute cellule cancéreuse présente le même défaut et la même cible exploitable.
⇪ Nouveaux traitements
Travis Christofferson (2014N37) évoque de nombreuses tentatives infructueuses d’utiliser de nouvelles molécules — ou combinaisons de molécules — pour le traitement du cancer par chimiothérapie. Dans un premier temps, le médicament paraît efficace, même au prix de souffrances causées par ses effets indésirables sur les cellules saines. Mais les rechutes fatales à court et moyen terme se révélent fréquentes. Les personnes qui ont survécu à des cancers localisés suite à un traitement par chimiothérapie sont aussi exposées à un risque nettement accru (2 à 10 fois) de maladies cardiovasculaires, effet indésirable des médicaments (Strongman H et al., 2019N60).
Cet avis mérite d’être relativisé au vu des statistiques de mortalité par cancer. Selon les données de la société américaine de cancérologie publiées dans CA : Cancer Journal for Clinicians, en 25 ans la mortalité par cancer a diminué de 27 % aux États-Unis (Le Quotidien du Médecin, 9/1/2019N61) :
La société américaine de cancérologie attribue cette amélioration au recul du tabagisme d’une part, et aux progrès réalisés dans la détection précoce et le traitement de 4 cancers les plus fréquents – poumon, sein, prostate et côlon – d’autre part. Le taux de décès lié au cancer du poumon a ainsi chuté de 48 % entre 1990 et 2016 chez les hommes et de 23 % entre 2002 et 2016 chez les femmes. Le taux de décès lié au cancer du sein a diminué de 40 % entre 1989 et 2016, tandis que les taux de mortalité par cancer de la prostate et par cancer colorectal ont été réduits de moitié entre 1970 et 2016.
Dans le même temps, on souligne une augmentation de la mortalité d’autres cancers plus rares. C’est le cas du taux de mortalité du cancer du foie (+1,2 % par an entre 2012 et 2016 chez les hommes, +2,6 % chez les femmes) et du cancer du pancréas (+0,3 % par an chez les hommes).
La probabilité de survie d’un patient dans une période donnée est évaluée à l’aide de la formule de Kaplan-Meier (voir discussionN62) :
(nbre de patients vivants au départ – nbre de patients décédés) / nbre de patients vivants au départ
Thomas Seyfried (voir vidéoN52 36:20) cite l’étude de Stupp R. et al. (2009N63) qui donne le résultat, pour 573 patients atteints de de glioblastome (N64 tumeur du cerveau), d’un traitement par chimiothérapie (témozolomide, TMZN65) combiné avec la radiothérapie, en comparaison avec la radiothérapie seule. Une « preuve » de l’efficacité du TMZ est établie par la différence entre la courbe bleue et la courbe rouge (figure ci-dessous).
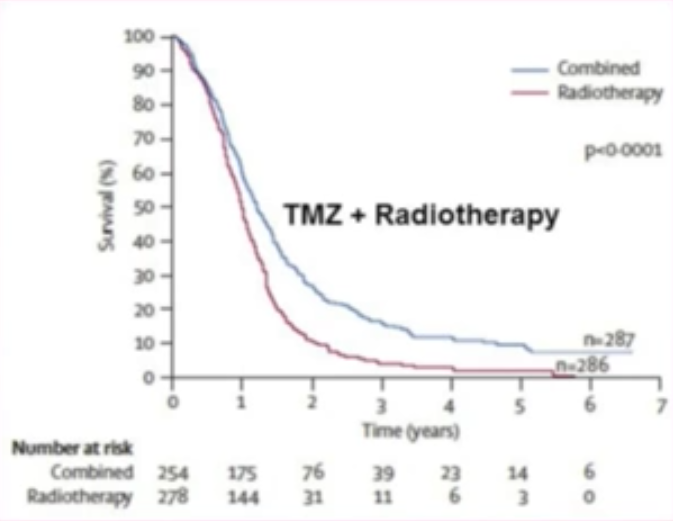
Les courbes illustrent toutefois le fait que 89% des patients étaient décédés au bout de 5 ans dans le groupe chimio+radiothérapie, et 97% dans le groupe radiothérapie seule. Ce qu’elles ne montrent pas est l’inconfort des patients pendant le temps du traitement — voir les effets indésirables en monothérapie du TMZN65 — ni le coût du médicament : pour le TMZ seul, environ 50 000 $ par année de traitement (voir sourceN66).
Enfin, l’étude de Brett E Johnson et al. (2014N67) révèle que les tumeurs apparaissant lors de récurrences présentent de nouvelles mutations qui ont été provoquées par le traitement au TMZ. On peut se demander, dans ce cas, pourquoi la survie des patients a légèrement augmenté. Seyfried suggère que ce pourrait être le seul effet de la restriction calorique dont les patients font l’expérience en raison des violents effets indésirables (vomissements etc.). Si cette hypothèse est vérifiée, l’efficacité (très relative) d’un médicament aussi coûteux serait uniquement liée à ses effets secondaires…
Christofferson (2014N37, p. 157) écrit :
Depuis que l’HerceptineN68 [GenentechN69 1998] a initié la révolution des médicaments ciblés, un regard objectif sur les résultats dresse un tableau déprimant. « Une estimation prudente du nombre de thérapies ciblées testées chez des patients atteints de cancer dans la dernière décennie a été sept cent », a déclaré Antonio Tito Fojo, Ph.D., chef de la Section de thérapeutique expérimentale et chercheur principal des Medical Oncology Branch Affiliates au Center for Cancer Research du National Cancer Institute à Bethesda, Maryland, « cependant aucun patient atteint de tumeur solide n’a été guéri par thérapie ciblée sans cette période. Le nombre de thérapies ciblées qui ont prolongé la survie d’un an, par rapport à un traitement conventionnel, est zéro ». [N70]
L’écrivain scientifique Ralph Moss a signalé les critères bizarres que la FDA [Food and Drug Administration aux USA] utilise pour approuver [la mise sur le marché des] médicaments, permettant à une foule de médicaments inefficaces d’obtenir l’approbation :
« Si vous pouvez réduire la tumeur de 50 pour cent ou plus pendant 28 jours vous avez la définition de la FDA d’un médicament actif. On appelle cela un taux de réponse, de sorte que vous avez une réponse… (Mais) quand vous cherchez s’il y a une quelconque prolongation de la vie après ce traitement, ce que vous trouvez est toutes sortes d’abracadabra et de pirouettes sur la survie sans maladie, et ceci et cela. »
[…]
Un exemple de l’état actuel de médicaments contre le cancer est le bevacizumab (N71 Avastin). Il a reçu l’approbation de la FDA en 2004 pour le cancer du côlon métastatique et plus tard a reçu l’approbation pour d’autres applications, y compris le cancer du sein. Traiter le patient moyen de cancer du sein avec l’Avastin coûte 90 816 dollars par an, sans prolonger la survie globale. Mais comme il a diminué des tumeurs dans quelques cas, la FDA l’a approuvé, ce qui souligne les critères absurdes utilisés pour l’approbation des médicaments. Pire, les patients qui ont été traités par l’Avastin en complément du paclitaxelN72 avaient deux fois plus de risque de subir une toxicité nettement plus élevée.
 Précisons que la FDA ne mandate pas d’organisme indépendant pour effectuer des tests de médicaments. Elle se base uniquement sur les rapports fournis par les industriels pour approuver la mise sur le marché de leurs produits. Le scandale du VioxxN73 diffusé par Merck peut donner à réfléchir (voir articleN74)…
Précisons que la FDA ne mandate pas d’organisme indépendant pour effectuer des tests de médicaments. Elle se base uniquement sur les rapports fournis par les industriels pour approuver la mise sur le marché de leurs produits. Le scandale du VioxxN73 diffusé par Merck peut donner à réfléchir (voir articleN74)…
La découverte de molécules susceptibles d’améliorer considérablement le soin de cancers agressifs fait l’objet de campagnes de communication qui facilitent la levée de fonds mais peuvent donner aux malades de faux espoirs : soit la molécule, testée sur des cultures de cellules ou en expérimentation animale, se révèlera inefficace pour le traitement des humains, soit les essais cliniques permettant d’aboutir à une posologie et à son autorisation de mise sur le marché prendront de longues années au terme desquelles les patients ciblés seront décédés. Un exemple d’annonce faussement prometteuse est celui, en 2019, de l’hydrochloride PJ34 « qui soigne en 14 jours le cancer du pancréas » mais seulement pour une souche de souris de laboratoireN75…
Eléonore Djikeussi écrit (2022N76 p. 32) :
Le schéma se répète : une ère nouvelle est annoncée, mais la victoire n’est jamais au rendez-vous.
L’approche de cette guerre ciblée, de précision chimique, connaît un engouement tel que de nouvelles molécules arrivent sur le marché à un rythme qui ne permet plus à un oncologue même chevronnée d’en étudier la littérature scientifique et de manipuler l’ensemble avec expertise.
Après la brève carrière de l’Herceptine, un médicament nommé GLIVEC — ou GLEEVEC — (mésilate d’imatinibN77) a suscité d’immenses espoirs. Il ciblait une mutation particulière rencontrée dans une forme rare de leucémie : la leucémie myéloïde chronique (CMLN78). Ce médicament était le premier à guérir le cancer en empêchant une mutation, justifiant par cela la théorie génétique. Harold Varmus a publié à cette occasion un essai titré The New Era in Cancer Research (La nouvelle ère de la recherche sur le cancer). On évoquait en effet un « changement de paradigme » dans le développement des médicaments contre le cancer.
Le GLIVEC s’attaque à un chromosome défectueux, surnommé “the Philadelphia chromosome”, découvert en 1960 par Peter Nowel dans un laboratoire de Philadelphie. La découverte du traitement est attribuée à Jurg Zimmermann et Nicholas Lydon dans un laboratoire suisse (Ciba-Geigy). Lydon a réussi à mener le test clinique après un retard considérable causé par la fusion de Ciba-Geigy avec Sandoz pour former Novartis, dont les dirigeants refusaient de prendre le risque d’un financement de 100 millions de dollars sans garantie de retour sur investissement, car la maladie ciblée avait une incidence rare… Le test a finalement eu lieu avec la collaboration de Brian Druker qui travaillait au Dana Farber Cancer Institute de Boston. Sur 54 patients, 53 ont réagi positivement quelques jours après avoir commencé le traitement. Les patients retrouvaient totalement la santé et leur vie redevenait normale.
Le succès du GLIVEC était total (voir Pray L, 2008N79) bien qu’il ne puisse soigner qu’une forme rare de cancer (la CML). Mais sa pertinence en tant que preuve de la théorie génétique n’a pas été confirmée. D’une part, la CML procède d’une mutation unique et toujours identique. D’autre part, certaines personnes possédant cette mutation ne développent pas de cancer. Enfin, environ 20% des personnes souffrant de CML à un stade avancé ne survivent pas malgré le traitement. La mutation, en soi, ne peut donc pas être considérée comme la cause première du cancer. Seyfried et Pedersen ont d’ailleurs remarqué que l’action du GLIVEC apportait de l’eau au moulin de la théorie métabolique — voir Seyfried TN et al. (2014N80).
Eléonore Djikeussi précise toutefois (2022N76 p. 32–33) :
Un des heurs de l’Iminatib, Glivec, est peut-être qu’il interfère avec le métabolisme glucidique, activé dans les cellules malignes par un mécanisme qui implique PI3KN81/AktN82 […], bloquant ainsi l’avidité de ces cellules pour le glucose, leur carburant principal, nécessaire pour leur production d’énergie et la fabrication de molécules servant à produire de nouvelles cellulees malignes.
⇪ L’histoire mouvementée du 3BP
Le coût annuel des médicaments cancéreux, aux USA, est passé d’environ 5000 $ avant 2000 à 40 000 $ vers 2005, et en 2012 presque chaque nouveau médicament coûtait plus de 100 000 $ (Christofferson T, 2014N37, p. 158). C’est le cas notamment du GLIVEC, alors que l’Herceptine ne coûtait « que » 70 000 $ (Nordqvist C, 2021N83). Dans cette configuration, la découverte d’un médicament très peu coûteux, et capable d’agir sélectivement sur tous les types de cellules cancéreuses, constituerait une avancée significative. C’est le cas du 3‑Bromopyruvate (3BP) (voir Pedersen P, 2012N84 et Valenti D et al., 2015N85) dont l’histoire a été tumulteuse. On peut en lire les épisodes dans l’ouvrage de Christofferson (2014N37, p. 104–123) basé sur des entretiens avec les principaux protagonistes, notamment Young H. Ko et Peter L. Pedersen.
Young Hee Ko a été formée jusqu’en 1981 à l’Université Kon-Kuk à Séoul (Corée du Sud). L’année suivante, elle a émigré aux États-Unis et s’est inscrite au programme de master de physiologie de la nutrition à l’Université de l’État de l’Iowa. Munie de son diplôme, en 1985, elle a poursuivi ses études jusqu’au doctorat de biochimie à l’Université de l’État de Washington (en 1990). Elle a ensuite rejoint pour un post-doc le laboratoire de Peter Pedersen à l’Université Johns Hopkins de Baltimore, où elle s’est intéressée à la fibrose kystique (mucoviscidoseN86), réussissant à identifier le codon défectueux qui altère la protéine CFTR (cystic fibrosis transmembrane conductance regulator). Elle a été ensuite orientée vers la recherche sur le cancer, toujours sous la direction de Pederson qui explorait comme fondation théorique l’hypothèse de WarburgN38 à contre-courant de la théorie génétique (SMT, Somatic Mutation Theory). Il s’agissait pour eux de trouver un moyen d’isoler et inhiber les effets de l’hexokinase IIN87, une enzyme dont l’expression est accrue dans de nombreux cancers (Mathupala SP et al., 2006N88).
Ko est partie du constat que les cellules cancéreuses produisent de l’acide lactique en très grande quantité, de sorte qu’elles pourraient mourir asphyxiées si on les empêchait d’en éliminer l’excès. Ces cellules surproduisent dans leur membrane une protéine qu’on appelle transporteur de monocarboxylate (MCTN89), agissant comme une porte qui autorise l’acide lactique et l’anion pyruvateN90 à entrer et sortir de la cellule. Les « portes » sont plus nombreuses dans les cellules cancéreuses que dans les cellules saines.

(Szalka Petriego)
CC BY-SA 3.0
La chercheuse s’est souvenue qu’elle avait travaillé à Washington avec le 3‑Bromopyruvate (3BP), une molécule dont la structure est très proche de celle du pyruvate. La protéine MCT pourrait donc la laisser passer, de sorte qu’une fois parvenue à l’intérieur de la cellule cancéreuse elle se comporterait comme un cheval de Troie en détruisant de l’hexokinase IIN87. La méthode paraissait trop simple pour être efficace, mais Young Ko a tenu à la vérifier. Elle a cultivé des cellules cancéreuses dans une boîte de Pétri et observé leurs réactions à diverses molécules, en comparaison avec le 3BP, découvrant que cette dernière molécule agissait bien plus efficacement, et sur tous les types de cancer, ce qui n’était pas le cas des autres molécules. La différence majeure tenait au fait que le médicament intervient sur un aspect invariant du métabolisme des cellules plutôt que de cibler des configurations particulières de mutations.
Par la suite, Ko et Pedersen ont vérifié l’efficacité du 3BP en expérimentation animale, avec le même succès, constatant que la molécule n’induisait pas d’effets secondaires qui auraient eu une issue fatale, comme ils le craignaient au départ. Il restait à passer à l’expérimentation humaine, une étape qui s’est révélée extrêmement difficile et douloureuse pour les chercheurs, pour plusieurs raisons. Une molécule qui réduisait à une centaine de dollars le coût du traitement d’un cancer n’intéressait pas particulièrement l’industrie pharmaceutique, principal financeur, aux USA, d’essais cliniques qui se comptent en millions de dollars. Mais la promesse d’un succès scientifique aiguisait les appétits, entre autres celui d’un collaborateur subalterne que les découvreurs avaient associé au dépôt de leur premier brevet sur le traitement anticancéreux par 3BP — « la plus grossière erreur de ma carrière » déclarait Pederson (Christofferson T, 2014N37, p. 107). Cet ancien collaborateur a fondé par la suite un laboratoire concurrent (PreScience LabsN91).
Young Ko est entrée en conflit avec le département de radiologie de Johns Hopkins qui lui avait offert un contrat de trois ans sans mettre un espace à sa disposition pour mener son expérimentation (Christofferson T, 2014N37, p. 104–107). Elle pouvait demander des subventions mais n’avait aucune chance de les obtenir, faute d’espace de travail. La situation s’est envenimée après l’octroi d’une bourse Susan B. Komen et que l’espace promis lui ait encore été refusé par Chi Dang, vice-doyen de la recherche à Johns Hopkins. Quand Pedersen avait présenté les perspectives du 3BP dans le traitement du cancer, le laboratoire avait immédiatement commandé un stock de 3BP pour son usage, puis obtenu de Ko plus de 80 heures de transfert d’expertise… Ayant protesté contre ce qu’elle percevait comme une appropriation de son travail et un sabotage de sa carrière, elle a été licenciée après avoir refusé une expertise psychiatrique exigée par son employeur. Elle a déposé une plainte pour discrimination le 1er juin 2005 à la cour du District de Maryland, qui a été rejetée (voir motifs : #1:05-cv-01475-WDQN92).
La recherche d’un financement pour un essai clinique sur des patients du cancer s’est avérée un véritable parcours du combattant, certains laboratoires concurrents essayant de s’emparer de la technique en soutirant un maximum d’informations des auteurs — et de leurs dossiers d’expertise…
Le premier essai du 3BP sur un humain a eu lieu en 2008, lorsque Young Ko a été contactée par le père d’Yvar Verhoeven, un enfant néerlandais de 16 ans atteint d’un carcinome hépatocellulaireN93 dont 95% du foie s’était consumé en produisant des masses cancéreuses de la grosseur du poing. Le cancer avait déjà migré vers son cœur. Après une longue procédure pour trouver un médecin acceptant d’essayer le traitement, puis pour obtenir le feu vert du comité d’éthique, l’intervention a été menée fin février 2009 par Thomas Vogl, de l’Université de Francfort. La guérison de ce patient a été rapide et complète, sans manifestation d’effets secondaires. Un an plus tard, toutefois, il a succombé à une pneumonie en raison de l’impossibilité de lui administrer des antibiotiques qu’un foie réduit à 5% de son volume n’aurait pas pu supporter. L’examen post-mortem a confirmé qu’aucune cellule cancéreuse ne subsistait dans son organisme, ce qui écartait tout soupçon de rechute.
L’expérimentation avec le 3BP se poursuit. Cette molécule est une de celles qui offrent le plus de perspectives pour le traitement du cancer (voir Ko YH et al., 2012N94, El Sayed SM et al., 2014N95 etc.). Elle bénéficie d’une validation « technique » en expérimentation animale, mais il faudrait réunir 3 millions de dollars pour procéder à une validation clinique en expérimentation humaine. Même PreScience LabsN91 qui a reçu en 2013 l’autorisation de la US Food & Drug Administration (FDAN96) (voir communiqué de presseN97) n’a pas encore obtenu, à ce jour, un financement de son premier essai clinique (phase 1).
⇪ Autres traitements récents
De nouveaux traitements anti-cancéreux sont apparus : immunothérapies anticancéreuses et inhibiteurs de tyrosines kinases. Ils sont mieux ciblés et donc plus efficaces, mais ils entraînent aussi de nouveaux effets secondaires qui demandent un suivi particulier. D’après Le Quotidien du médecin (29/11/2016N98),
Sous chimiothérapies conventionnelles, des rashs cutanés [N99] peuvent être observés sous doxorubicine (par exemple), des paronychies [N100] (sous taxanes), des syndromes mains-pieds (sous fluoropyrimidine). Sous thérapies ciblées, xérose [N101], paronychies (hémorragies sous-unguéales, pseudo-panaris,…), pulpites sèches [N102], folliculites [N103], kératose pilaire [N104] (sous anti-BRAF, par exemple), rash, ulcérations cutanéo-muqueuses (par exemple, sous inhibiteurs de mTOR), syndromes mains-pieds (sous inhibiteurs de tyrosines kinases (TKI)) etc… sont fréquentes et doivent être anticipées par des mesures préventives adaptées pour limiter les problèmes liés à la mauvaise observance.
La thérapie d’acidification photodynamique utilise l’injection d’une simple dose de nitrobenzaldéhydeN105 suivie d’une exposition à un rayonnement ultraviolet, qui a pour effet d’acidifier les cellules cancéreuses et de déclencher leur apoptoseN21. Elle a été vérifiée sur des souris atteintes de la forme la plus invasive de cancer du sein (triple négatif) : en peu de temps, la croissance de la tumeur était interrompue, sans que les cellules saines ne soient affectées, et le taux de survie après traitement était significativement augmenté (Kadri NB et al., 2016N106). Selon l’estimation d’un chercheur, 95% des cellules cancéreuses auraient été détruites en deux heures.
L’équipe essaie maintenant de développer une nanoparticule qui permettrait de cibler les cellules cancéreuses de cancers métastasés (University of Texas, 2016N107). L’enjeu est crucial car les métastases contribuent très fortement à la faible efficacité de la chimiothérapie conventionnelle, comme le souligne Peter Wise (2016N108) :
Une méta-analyse publiée en 2004, à partir d’essais randomisés australiens et américains, a exploré la contribution de la chimiothérapie cytotoxique à la survie à cinq ans chez 250 000 adultes atteints de cancers solides [métastasés]. Un effet important a été montré sur la survie à cinq ans seulement pour le cancer des testicules (40%), la maladie de Hodgkin (37%), le cancer du col de l’utérus (12%), le lymphome (10,5%) et le cancer de l’ovaire (8,8%). Ensemble, ils représentaient moins de 10% de tous les cas. Dans les 90% restant des patients, y compris ceux ayant les tumeurs les plus fréquentes du poumon, de la prostate, du colorectum et du sein, les traitement médicamenteux ont augmenté la survie à cinq ans de moins de 2,5%, soit un bénéfice global de survie d’environ trois mois.
De manière similaire, 14 nouveaux régimes thérapeutiques consécutifs pour les cancers solides adultes approuvés par l’Agence Européenne des Médicaments ont abouti à un bénéfice global moyen de survie de 1,2 mois par rapport aux régimes qui leur étaient comparés.
Les nouveaux médicaments n’ont pas été meilleurs : 48 nouveaux régimes approuvés par la Food and Drug Administration entre 2002 et 2014 offraient une médiane de 2,1 mois de bénéfice de survie globale.
Le traitement médicamenteux ne peut donc que partiellement expliquer l’amélioration de 20% dans la survie de cinq ans mentionnés ci-dessus. L’évolution du diagnostic et du traitement précoces peut y avoir contribué beaucoup plus.
Les travaux de Blasco MT et al. (2019N109, voir commentaire en français : N110) en expérimentation animale ouvrent une piste prometteuse pour la guérison de l’adénocarcinome du pancréas, une forme particulièrement agressive de cancer qui a touché plus de 14000 personnes en 2017 en France.
À signaler aussi — et la liste est loin d’être complète — la radio-immunothérapieN111 étudiée au Centre de Recherche en Cancérologie et Immunologie de Nantes-Angers (ARC, 2018N112) :
Le principe général de la radio-immunothérapie est d’associer une molécule radioactive à un anticorps qui, une fois injecté au patient, se fixe sur une protéine spécifiquement exposée à la surface des cellules cancéreuses. Les anticorps qui ne rencontrent pas leur cible sont progressivement éliminés par l’organisme et la radioactivité se concentre ainsi au niveau des cellules tumorales, ainsi exposées à l’irradiation. Cette technique, déjà validée en seconde ligne dans la prise en charge de lymphomes non-hodgkiniens, a comme principal avantage de toucher potentiellement toutes les cellules cancéreuses, y compris celles qui ne sont pas localisées au niveau de la tumeur primaire : contrairement à l’irradiation locale d’une radiothérapie classique, la radio-immunothérapie permet d’exposer les cellules tumorales qui circulent dans le sang ou les micro-métastases, invisibles à l’imagerie.
Selon François Davodeau, « Cette technique pourrait donc offrir la possibilité de détruire la tumeur mais aussi les métastases, y compris celles que l’on n’a pas encore décelées, ainsi que les cellules tumorales qui peuvent demeurer à la suite d’une chirurgie d’exérèse1 de la tumeur, responsables de la récidive du cancer. »
À l’Université de Cardiff, une équipe a publié une étude découvrant la découverte d’une cellule immunitaite capable de s’attaquer à tous les types de cancer (Crowther MD et al, 2020N113) :
Le ciblage des cellules cancéreuses induit par les cellules T et indépendant de l’antigène leucocytaire humain (HLA) permettrait la destruction immunitaire des tumeurs malignes chez tous les individus. Ici, nous utilisons le criblage CRISPR-Cas9 [N114] à l’échelle du génome pour établir qu’un récepteur des cellules T (TCR) a reconnu et tué la plupart des types de cancer humain via la protéine monomorphe liée au CMH de classe I, MR1, tout en restant inerte vis-à-vis des cellules non cancéreuses. Contrairement aux cellules T invariantes associées aux muqueuses, la reconnaissance des cellules cibles par le TCR était indépendante de la charge bactérienne. De plus, l’ajout en fonction de la concentration des ligands métabolites liés à la vitamine B de MR1 a réduit la reconnaissance TCR des cellules cancéreuses, ce qui suggère que la reconnaissance s’est produite via la détection du métabolome [N115] du cancer. Un clone de cellules T restreint par MR1 a induit une régression in vivo de la leucémie et a conféré une survie accrue des souris NSG. Le transfert de TCR aux cellules T des patients a permis de tuer le mélanome autologue et non autologue. Ces résultats ouvrent des perspectives aux immunothérapies indépendantes du HLA, pan-cancéreuses et pan-populationnelles.
L’accès rapide aux nouveaux traitements n’est pas toujours un bénéfice pour les patients. La publication en 2019 de données allemandes sur les médicaments mis sur le marché a montré moins de 25 % des anticancéreux avaient des effets sur la survie globale (Maisonneuve H, 2019N116).
Dans son article sur « … les manipulations des firmes pour favoriser leurs traitements » (2021N117), Jérémy Anso dénonce « l’utilisation de soin de référence non optimal dans les groupes contrôles, qui favorisent les traitements des firmes », cette pratique ayant tendance à se généraliser dans la recherche en oncologie. Citant à plusieurs reprises le chercheur Vinay Prasad (Mohyuddin RM et al., 2021N118) il écrit notamment :
L’équipe de chercheurs avec Vinay Prasad a montré que sur les 49 études cliniques menées sur le myélome multiple :
• 14 % ont utilisé des traitements de référence (soin standard) dépassés dans les groupes contrôles et moins efficaces connus avant la conduite de l’étude ;
• 18 % ont continué d’utiliser des soins standards sous-optimaux malgré la publication de nouvelle référence médicale, sans modification du groupe contrôle.
Autrement dit, ces groupes contrôles étaient par nature défavorisés avec des soins standards sous-optimaux qui peuvent artificiellement gonfler l’efficacité des nouvelles thérapies étudiées.
[…]
On remarque donc bien une généralisation de l’utilisation des raccourcis et des critères substitutifs en oncologie avec le risque de voir autoriser des thérapies qui n’apportent aucun bénéfice pour les malades, en condition réelle d’utilisation.
⇪ Toxines de Coley : un ancêtre de l’immunothérapie ?
L’histoire du traitement par les toxines de ColeyN119 est instructive. Non qu’il s’agisse d’un nouveau traitement, puisqu’il a été inventé à la fin du 19e siècle, mais parce qu’elle illustre les aléas de la recherche médicale.
Ce traitement consistait à injecter au patient un mélange de bactéries tuées par la chaleur afin de stimuler leurs « défenses naturelles »N119. Chirurgien, William ColeyN120 avait eu cette idée en remarquant que des patients qui avaient une infection cutanée après avoir été opérés d’un cancer voyaient parfois leur cancer régresser.
Matthew Tontonoz (2015N121) raconte que ce traitement avait montré son efficacité sur plus de 2000 cas à une époque précédant la découverte des rayons X en 1895 et leur utilisation pour le traitement des cancers, remplacée peu après par le traitement au radium. Contrairement aux toxines de Coley, la radiothérapie affichait des résultats clairs et cohérents chez presque tous les patientsN121. D’autre part, le mécanisme du traitement de Coley n’était pas connu. Après le décès de Coley, sa fille Helen Coley Nauts, qui n’avait aucune formation scientifique, a incité des scientifiques à poursuivre leurs recherches sur ce traitement. Elle avait pour cela compilé les notes de son père et entretenait une abondante correspondance avec les chercheurs de l’époque qui commençaient à travailler sur la chimiothérapie. Mais elle faisait aussi preuve d’un enthousiasme débordant, entre autres dans ses échanges avec Cornelius Rhoads qui était une sommité de la recherche médicale. Après un soutien initial, ce dernier a fini par changer son fusil d’épaule, exprimant des réserves sur la validité des travaux de Coley.
À la fin de la guerre, en 1945, quand Helene Nauts l’a recontacté sans succès pour demander un financement et même un emploi dans l’équipe hospitalière qui effectuerait les travaux. Malgré (et à cause de) son insistance, elle a reçu de Rhoads, en 1950, une dernière réponse l’informant que toute son équipe était engagée sur la voie de la recherche en chimiothérapie. Refusant d’abandonner le projet malgré cet échec, elle a créé le Cancer Research Institute (CRI) en 1953. Elle a appris quelques années plus tard que Cornelius Rhoads était intervenu en coulisses pour l’empêcher d’obtenir un financement de John D. Rockefeller Jr. qui avait été un ami de son père. Rhoads a même ordonné en 1955 la fermeture du laboratoire qui fabriquait le traitement de Coley.
Aujourd’hui, on considère que le traitement par les toxines de Coley serait « à l’origine de l’immunothérapie » et que son inventeur était « en avance sur son temps ». Mais, faute d’explication sur son fonctionnement, cette thérapie avait été classée par l’American Cancer Society parmi les « thérapies du cancer non prouvées », sans réfuter l’intérêt de nouvelles recherches. Un autre obstacle est apparu avec le Drug Efficacy Amendment passé par le Congrès américain en 1962, qui obligeait tout médicament à prouver son efficacité pour âtre autorisé par la Food and Drug Administration. Le traitement de William Coley n’a pas bénéficié, en dépit de son ancienneté, de la même dérogation que d’autres médicaments « non prouvés » comme l’aspirine. La preuve de l’efficacité dans les règles de l’art est coûteuse et difficile, d’autant plus que les toxines de Coley sont préparées par le biais de bactéries dont les noms sont identifiés mais qui peuvent prendre de multiples formes. En 1976 un essai clinique a été interrompu malgré un effet prometteur sur le lymphome de Hodgkin parce que les statistiques de survie convergeaient sur le long terme. Aucun traitement similaire n’a passé le cap de la validation.
Ce sont les premiers travaux en immunologie du cancer, dans les années 1970, qui ont donné une impulsion nouvelle à la recherche sur les toxines de Coley en se focalisant sur le mécanisme du traitement. Tontonoz (2015N121) écrit :
Enfin, le moment semblait venu de revoir les toxines de Coley, avec une compréhension plus profonde du fonctionnement du système immunitaire. En 2007, le CRI a financé un essai clinique de phase I sur les toxines de Coley chez des patients atteints de divers types de cancer. L’essai a été réalisé à l’hôpital Krankenhaus Nordwest de Francfort, en Allemagne, sous la direction de Elke Jäger, M.D., membre du réseau d’essais cliniques du CRI. Les patients recevaient des injections sous-cutanées de toxines de Coley deux fois par semaine jusqu’à ce que la fièvre s’induise, puis quatre doses supplémentaires.
Contrairement aux précédents essais cliniques sur les toxines, celui-ci a été mené avec des toxines fabriquées conformément aux directives de bonne pratique clinique (GCP), avec des composants bactériens normalisés. L’essai incluait également des mesures de laboratoire des réponses immunitaires, par exemple des taux sanguins de cytokines, qui n’étaient pas possibles auparavant, et ciblait spécifiquement les patients dont les cancers exprimaient le drapeau moléculaire spécifique appelé NY-ESO‑1.
Comme toutes les études de phase I, l’objectif principal de cette étude n’était pas de déterminer l’efficacité clinique, mais d’en déterminer la sécurité et d’évaluer la posologie optimale. Néanmoins, il y a eu des résultats très prometteurs. Les toxines ont été efficaces pour induire de la fièvre et de fortes poussées de cytokines. Un patient atteint d’un cancer de la vessie métastatique a eu une réponse clinique claire au traitement, avec une réduction de 50% de son cancer qui était corrélée à des taux élevés de cytokines.
Toutefois, ce genre d’étude n’a pas pu être repris, à la fois par manque de financement et en raison des contraintes nouvelles imposées par les comités d’éthique de la recherche clinique mis en œuvre dans les hôpitaux. Entre autres, la preuve de l’efficacité des toxines de Coley était une élévation de température chez les patients qui pouvait laisser croire à une détérioration de leur état, et pendant l’essai cette fièvre pouvait persister pendant des semaines ou des mois à mesure que les doses étaient augmentées.
Il est possible que les nouveaux traitements en immunothérapie aient dépassé et rendu obsolète l’approche de William Coley. Matthew Tontonoz (2015N121) conclut :
Coley avait lui-même constaté que les toxines étaient plus efficaces pour prévenir les récidives lorsqu’elles étaient administrées après une intervention chirurgicale. Cela reflète les approches actuelles d’immunothérapie associant chirurgie et vaccins anticancéreux.
⇪ Cellules souches
Une thérapie nouvelle a été annoncée en 2012 avec un forte médiatisation en Italie. Elle consisterait à extraire des cellules souches de la moelle épinière du patient puis à les « traiter » afin qu’elles se différencient en cellules nerveuses. (Sans intervention, ces cellules souches ne peuvent se différencier qu’en cellules osseuses, graisseuses ou cartilagineuses.) Réinjectées dans l’organisme elles permettraient sa guérison.
Cette méthode StaminaN122 est issue d’un laboratoire de Brescia dirigé par Davide Vannoni, ancien professeur de psychologie de l’Université d’Udine devenu homme d’affaires. Aucune preuve scientifique de sa faisabilité, de son efficacité et de son innocuité n’a été publiée. La revue Nature a révélé qu’une micrographie essentielle dans la demande de brevet, décrivant deux cellules nerveuses apparemment différenciées des cellules stromales de la moelle osseuse, avait été copiée d’un document de recherche publié en 2003 par une équipe russe et ukrainienneN123 — ce qui a été confirmé par Elena Schegelskaya, une co-auteure du document plagié.
En 2015, Vannoni a été reconnu coupable d’accusations criminelles liées à l’administration d’un traitement non prouvé et interdit d’exercer une profession médicale en Italie.
Les publications frauduleuses annonçant le succès de nouveaux médicaments contre le cancer sont nombreuses (voir par exempleN124) étant donné l’impact médiatique/économique de ces annonces et leurs retombées en termes d’avancement de carrière.
⇪ Thérapie génique
Le fait que le projet TCGA n’ait pas (sauf rares exceptions) comblé l’espoir d’identifier — et par la suite modifier — des schémas invariants de mutations génétiques caractérisant les diverses formes de cancer, ne signifie pas pour autant qu’aucune thérapie géniqueN125 ne pourrait produire de résultat.
On écoutera avec beaucoup d’intérêt un entretien avec Alain FischerN126, chercheur en biologie à l’hôpital Necker (Paris), dont l’équipe a mis au point, dans les années 2000, un traitement génique des enfants-bullesN127 atteints d’immunodéficience sévère (émission Révolutions médicales, 26/01/2016N128). La thérapie consistait à intervenir sur un gène sujet à une mutation indésirable qui empêchait la production de lymphocytes TN129 dans l’organisme, privant l’organisme de ces enfants de l’essentiel de leurs défenses immunitaires (Cavazzana-Calvo M et al., 2000N130). Pour cela, on a transféré dans leurs cellules sanguines, par le biais d’un rétrovirus, un gène fonctionnel restaurant la fonctionnalité du récepteur à l’interleukine 2N131.
Malgré un grave effet secondaire — le déclenchement, chez certains patients, de leucémies induites par l’activation indésirée d’oncogènesN23 — la technique a pu être corrigée pour donner satisfaction en minimisant les risques. Elle a été appliquée avec succès sur quelques patients atteints de maladies rares, notamment pour le traitement de lymphome (N132 cancer des nœuds lymphatiques) par le transfert de gènes codants pour des molécules artificielles capables de rediriger des lymphocytes TN129 à l’encontre des cellules leucémiques (WikipediaN125).
On peut donc s’attendre à quelques avancées dans le domaine du cancer, non pas pour intervenir sur des mutations qui dans la majorité des cas, nous l’avons vu, sont trop complexes et imprévisibles, mais plutôt pour modifier le code génétique de cellules utilisées par le système immunitaire en vue de les programmer à la destruction de cellules malignes, ou encore de lever les obstacles chimiques qui empêchent les lymphocytes d’attaquer les cellules cancéreuses. Cette dernière approche très prometteuse a été présentée par Caroline Robert et Éric Vivier dans une émission de la série Matières à penser (février 2019N133). Une des difficultés de l’immunothérapie est le coup très élevé du traitement — environ 80 000 euros selon les intervenants de cette émission — et l’incertitude encore très grande de son efficacité pour un patient en particulier.
⇪ Chronothérapie
L’équipe U935 de l’INSERMN134 s’intéresse à l’optimalisation de la chronothérapieN135 des cancers et de la fonction hépatique post-opératoire. Cette branche de la chronobiologieN136 a pour but de synchroniser les rythmes circadiens et de les utiliser à des fins thérapeutiques. Annabelle Ballesta, chargée de la modélisation mathématique dans le cadre de l’équipe ATIP Avenir, écritN137 :
Je développe des modèles mathématiques fondés sur la physiologie cellulaire : les données de la littérature et les travaux spécifiques conduits pour l’occasion dans le domaine de la chronobiologie permettent de créer un modèle virtuel cellulaire, animal, puis humain qui vise à reproduire in silico ce qui se passe biologiquement. Les paramètres que nous y intégrons sont ajustés aux données biologiques pour assurer la validité du modèle. Une fois optimisés, ils permettent de modéliser l’impact d’un traitement selon le moment de la journée auquel il est administré. Ces modèles intègrent des réseaux de gènes ou de protéines qui sont décrits comme interagissant avec les médicaments étudiés, comme par exemple ceux qui régissent les processus de réparation de l’ADN, le cycle cellulaire ou la mort cellulaire… L’apport des mathématiques est de réduire le nombre d’expériences à mener en testant in silico un grand nombre d’hypothèses et en ne testant expérimentalement que celles qui s’avèrent pertinentes pour développer les connaissances biologiques.
[…]
Grâce à ce programme, nous menons de front deux projets : le premier consiste à étudier la façon dont la chronopharmacologie de trois médicaments prescrits dans les cancers digestifs est influencée par des facteurs comme l’âge, le sexe ou encore le chronotype des personnes — lève-tôt ou couche-tard. Le second vise à développer des stratégies personnalisées à partir de combinaisons de médicaments dits « ciblés », qui n’interagissent qu’avec une ou deux protéines intracellulaires, afin d’améliorer l’efficacité antitumorale du traitement. Ces deux thématiques se rejoignent dans leur finalité : individualiser les traitements selon la nature de la tumeur mais aussi selon le patient lui-même.
(Suite sur la page Cancer - dépistage)
Les références bibliographiques complètes sont sur la page Cancer - conclusion et références.
Le contenu de cet article ne se substitue pas aux recommandations des professionnels de santé consultés par les lecteurs.
▷ Liens
 Notes pour la version papier :
Notes pour la version papier :
- Les identifiants de liens permettent d’atteindre facilement les pages web auxquelles ils font référence.
- Pour visiter « 0bim », entrer dans un navigateur l’adresse « https://leti.lt/0bim ».
- On peut aussi consulter le serveur de liens https://leti.lt/liens et la liste des pages cibles https://leti.lt/liste.
- N1 · 52rn · James Dewey Watson – Wikipedia
- N2 · g4m1 · Rosalind Franklin – Wikipedia
- N3 · v7fv · Watson, James (2014). Type 2 diabetes as a redox disease. The Lancet, 383, 9919 : 841–843.
- N4 · jdt9 · Dérivé réactif de l’oxygène (ROS) – Wikipedia
- N5 · w6ci · Entraînement fractionné de haute intensité – HIIT – Wikipedia
- N6 · rvln · Nobelist James Watson proposes an unconventional view of type 2 diabetes causation
- N7 · zofl · Pancréas – Wikipedia
- N8 · a3u9 · Diabète de type 2 – Wikipedia
- N10 · 8zhv · Réticulum endoplasmique – Wikipedia
- N11 · ab2l · Pont disulfure – Wikipedia
- N12 · b8o4 · Repliement des protéines – Wikipedia
- N13 · kcld · Ristow, A et al. (2009). Antioxidants prevent health-promoting effects of physical exercise in humans. PNAS, 106, 21 : 8665–8670.
- N14 · 81fq · Hormèse – Wikipedia
- N15 · skr4 · Ristow M, Schmeisser K (2014). Mitohormesis : Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response. 12, 2:288–341. doi:10.2203/dose-response.13–035
- N16 · jvwz · Watson, James (2013). Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol 3 : 120144.
- N17 · 6qeo · Adénosine triphosphate (ATP) – Wikipedia
- N18 · gt0v · Carcinome – Wikipedia
- N19 · apqt · Transition épithélio-mésenchymateuse – Wikipedia
- N20 · 0ubw · Métastase – Wikipedia
- N21 · e3yx · Apoptose – Wikipedia
- N22 · vczo · Acétylcystéine – Wikipedia
- N23 · 12ty · Oncogène – Wikipedia
- N24 · dc13 · Glutathion – Wikipedia
- N25 · 9uvt · Superoxyde dismutase – Wikipedia
- N26 · 34fv · Catalase – Wikipedia
- N27 · xuml · Thiorédoxine – Wikipedia
- N28 · q81n · NFE2L2 – Nrf2 – Wikipedia
- N29 · odmk · IGF‑1 – Wikipedia
- N30 · ot3s · Velloso CP (2008). Regulation of muscle mass by growth hormone and IGF‑I. Br J Pharmacol. 154, 3 : 557–568. doi:10.1038/bjp.2008.153
- N31 · 8it2 · James Watson : Basing ‘war on cancer’ on genome research diverts resources
- N32 · 8i3j · Interférence par ARN – Wikipedia
- N33 · 5fyj · Kaelin G (2012). Use and Abuse of RNAi to Study Mammalian Gene Function. Science, 337, 6093 : 421–422. doi:10.1126/science.1225787
- N34 · jylh · Cold Spring Harbor Laboratory
- N35 · rh42 · Watson, James (2009). To Fight Cancer, Know the Enemy. The Opinion Pages, The New York Times, Aug. 5.
- N36 · e5j3 · AGIOS company
- N37 · rtjb · Christofferson, T (2014). Tripping Over the Truth : The Return of the Metabolic Theory of Cancer Illuminates a New and Hopeful Path to a Cure. Auto-édition, CreateSpace.
- N38 · 16ls · Warburg hypothesis – Wikipedia
- N39 · naqt · Émission de radio “Rosalind Franklin, à 2 brins du Nobel” – France Culture
- N40 · juu7 · Hanahan D & Weinberg RA (2000). The Hallmarks of Cancer. Cell, 100, 1 : 57–70.
- N41 · gvbs · Angiogenèse – Wikipedia
- N42 · 7yxa · Peter L. Pedersen, Ph.D.
- N43 · ll1a · Pedersen, Peter (2009). NCI and NIH Mitochondria Interest Group Seminar : Johns Hopkins’ Pedersen Addresses Role of Mitochondria in Cancer.
- N44 · mo8r · Warburg effect – Wikipedia
- N45 · eu51 · Hanahan D & Weinberg RA (2011). Hallmarks of cancer : the next generation. Cell, 144, 5 : 646–74.
- N46 · 6d8h · The hallmarks of cancer – Wikipedia
- N47 · alc0 · Mitochondrie – Wikipedia
- N48 · 66ps · Thomas N. Seyfried – author
- N49 · p5f7 · Thomas N. Seyfried – Academic Profile
- N50 · ygyn · McKinnell RG, Deggins BA, Labat DD. (1969). Transplantation of pluripotential nuclei from triploid frog tumors. Science. 165, 3891 : 394–6.
- N51 · vbou · Seyfried, TN (2015a). Cancer as a mitochondrial metabolic disease. Front Cell Dev Biol. 2015, 3 : 43.
- N52 · vatn · Seyfried, TN (2015b). Cancer : A Metabolic Disease With Metabolic Solutions. Institute for Human & Machine Cognition (IHMC). Youtube video.
- N53 · cz64 · O’Neill, Michael (2013). Review of “Cancer as a Metabolic Disease – Thomas Seyfried” – Update on metastasis. June 30. Blog article.
- N54 · 60vw · Macrophage – Wikipedia
- N55 · v36u · Leucocyte – Wikipedia
- N56 · afw2 · Cellule somatique – Wikipedia
- N57 · yy3w · Lazova, R et al. (2013). A Melanoma Brain Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation : First Evidence for Fusion in Human Cancer. PLoS ONE 8, 6 : e66731.
- N58 · pkyk · Cancer Scientists Prove Long-Standing Theory on How Cancer Spreads
- N59 · 3ovr · Pawelek, John (2014). Fusion of bone marrow-derived cells with cancer cells : metastasis as a secondary disease in cancer. Chin J Cancer. 2014, 33, 3 : 133–139.
- N60 · uyyz · Strongman, H et al. (2019). Medium and long-term risks of specific cardiovascular diseases in survivors of 20 adult cancers : a population-based cohort study using multiple linked UK electronic health records databases. The Lancet (on line). doi:10.1016/S0140-6736(19)31674–5
- N61 · q2gz · 2,6 millions de morts évités en 25 ans : incidence et mortalité par cancer en baisse aux États-Unis
- N62 · 4awr · Understanding survival analysis : Kaplan-Meier estimate
- N63 · lqsm · Stupp, R et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study : 5‑year analysis of the EORTC-NCIC trial. Lancet 10, 5 : 459–466.
- N64 · 6831 · Glioblastome multiforme – Wikipedia
- N65 · br8t · Témozolomide – Wikipedia
- N66 · h9h8 · Cost of temozolomide therapy and global care for recurrent malignant gliomas followed until death
- N67 · t15x · Johnson, BE et al. (2014). Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science, 343, 6167 : 189–93.
- N68 · jfa4 · Trastuzumab – Herceptine – Wikipedia
- N69 · cy9g · Genentech Company – Wikipedia
- N70 · xstp · Yin, Sandra (2011). Experts Question Benefits of High-Cost Cancer Care. Medscape, 5 décembre 2011.
- N71 · lik9 · Bévacizumab – Wikipedia
- N72 · t26p · Paclitaxel – Wikipedia
- N73 · mlqb · Rofécoxib – Vioxx – Wikipedia
- N74 · 6e45 · Merck débourse près d’un milliard de dollars pour solder le scandale Vioxx
- N75 · m1yb · Cancer du pancréas : un nouveau traitement pour soigner la maladie en 14 jours !
- N76 · l23i · Djikeussi, E (2022). Cancer : Maladie génétique ou crise énergétique cellulaire ? Le pouvoir de l’alimentation. Gap : Le Souffle d’Or.
- N77 · 62kq · Imatinib – Wikipedia
- N78 · tjwx · Leucémie myéloïde chronique – Wikipedia
- N79 · exfe · Pray, L. (2008). Gleevec : the Breakthrough in Cancer Treatment. Nature Education 1(1):37
- N80 · 2icq · Seyfried, TN (2014). Ketone Strong : Emerging evidence for a therapeutic role of ketone bodies in neurological and neurodegenerative diseases. The Journal of Lipid Research, 55, 1815–1817.
- N81 · abt3 · Phosphoinositide 3‑kinase – Wikipedia
- N82 · n732 · Akt1 – Wikipedia
- N83 · zf00 · Breast Cancer Cancer / Oncology One Year On Herceptin For Breast Cancer Ideal
- N84 · pg9k · Pedersen, Peter L. (2012). 3‑Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside : introduction to a special issue. J Bioenerg Biomembr. 2012, 44, 1 : 1–6.
- N85 · ki1i · Valenti, D et al. (2015). 3‑Bromopyruvate induces rapid human prostate cancer cell death by affecting cell energy metabolism, GSH pool and the glyoxalase system. J Bioenerg Biomembr. 2015, 47, 6 : 493–506.
- N86 · vrtp · Mucoviscidose – Wikipedia
- N87 · je9y · Hexokinase – Wikipedia
- N88 · gtrh · Mathupala, SP, Ko, YH, Pedersen, PL (2006). Hexokinase II : Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene,25, 34 : 4777–4786.
- N89 · qwnq · Monocarboxylate transporter – Wikipedia
- N90 · e87p · Acide pyruvique – Wikipedia
- N91 · d4jh · PreScience Labs
- N92 · j8vp · Case 1:05-cv-01475-WDQ Document 24 Filed 06/24/05 – PDF
- N93 · yked · Carcinome hépatocellulaire – Wikipedia
- N94 · juhz · Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL (2012). A translational study “case report” on the small molecule “energy blocker” 3‑bromopyruvate (3BP) as a potent anticancer agent : from bench side to bedside. J Bioenerg Biomembr. 44, 1 : 163–70.
- N95 · uana · El Sayed, SM et al. (2014). Safety and outcome of treatment of metastatic melanoma using 3‑bromopyruvate : a concise literature review and case study. Chin J Cancer. 2014, 33, 7 : 356–64.
- N96 · es2a · U.S. Food and Drug Administration
- N97 · wx61 · PreScience Labs Announced that the FDA Accepts IND Application for Novel Oncology Drug
- N98 · tlfr · Toxicité des traitements anti-cancéreux
- N99 · nrgd · Exanthème – Wikipedia
- N100 · bdeo · Paronychie
- N101 · vz9s · Xérose – Wikipedia
- N102 · tiuh · Pulpite – Wikipedia
- N103 · swh1 · Folliculite – Wikipedia
- N104 · 0jul · Kératose pilaire – Wikipedia
- N105 · z5u6 · Nitrobenzaldéhyde – Wikipedia
- N106 · hmcy · Kadri, Nuha Buchanan et al. (2016). Photodynamic acidification therapy to reduce triple negative breast cancer growth in vivo. Journal of Clinical Oncology, Vol 34, No 15_suppl (May 20 Supplement): e12574.
- N107 · 6krq · University of Texas at San Antonio (2016). New, non-invasive method developed to wipe out cancerous tumors : New treatment, requiring only a single dose and a beam of light, can kill up to 95 percent of cancer cells in two hours. ScienceDaily. ScienceDaily, 27 June.
- N108 · vscn · Wise, Peter (2016). Cancer drugs, survival, and ethics. BMJ 2016;355:i5792.
- N109 · sfm0 · Blasco, MT et al. (2019). Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C‑RAF. Cancer Cell, 35, 4 : 573–587. doi:10.1016/j.ccell.2019.03.002.
- N110 · f1ci · Cancer du pancréas : premier pas vers un traitement ?
- N111 · l9r9 · Radioimmunotherapy – Wikipedia
- N112 · s44l · ARC (2018). Cancers du sein triple-négatifs : les prémices d’une nouvelle stratégie. Communiqué, 19 juin.
- N113 · dkfu · Genome-wide CRISPR–Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I‑related protein MR1
- N114 · 5lkt · CRISPR-Cas9 – Wikipedia
- N115 · rkur · Métabolome – Wikipedia
- N116 · bz5d · Maisonneuve, H (2019). Les dysfonctionnements de la mise sur le marché des anticancéreux : l’accès rapide n’est pas un bénéfice pour les patients. Blog Rédaction Médicale et Scientifique.
- N117 · wlw3 · Anso, Jérémy (2021). Cancer : les manipulations des firmes pour favoriser leurs traitements. Blog Dur à Avaler.
- N118 · n3rh · Mohyuddin, RM et al. (2021). Quality of control groups in randomised trials of multiple myeloma enrolling in the USA : a systematic review. The Lancet Haematology, 8, 4 : e299-e30.
- N119 · 0o2s · Coley’s toxins – Wikipedia
- N120 · or28 · William Coley – Wikipedia
- N121 · 21n3 · Tontonoz, M (2015). What Ever Happened to Coley’s Toxins ? Article du site Cancer Research Institute (www.cancerresearch.org).
- N122 · da07 · Méthode Stamina – Wikipedia
- N123 · xqyz · Italian stem-cell trial based on flawed data
- N124 · 16uk · The wizard men curing breast cancer
- N125 · 2i8k · Thérapie génique – Wikipedia
- N126 · k5yf · Alain Fischer – Wikipedia
- N127 · 56e4 · Enfant-bulle – Wikipedia
- N128 · odvh · La thérapie génique sort-elle de la médecine expérimentale ?
- N129 · ee2n · Lymphocyte T – Wikipedia
- N130 · y60g · Cavazzana-Calvo, M et al. (2000). Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science, 288, 5466 : 669–672. doi:10.1126/science.288.5466.669
- N131 · wf2x · Interleukine 2 – Wikipedia
- N132 · qr1t · Lymphome – Wikipedia
- N133 · iv2n · L’immunothérapie du cancer – Émission Matières à penser, France Culture
- N134 · uo7e · INSERM U935 – Optimisation de la Chronothérapie des Cancers et de la Fonction Hépatique Post-opératoire
- N135 · r2zq · Chronothérapie – Wikipedia
- N136 · xodx · Chronobiologie – Wikipedia
- N137 · 7qwz · Annabelle Ballesta : Individualiser les traitements grâce aux maths et à la chronobiologie
Article créé le 20/11/2018 - modifié le 17/06/2024 à 14h54


